
分子相互作用理论计算
理论计算团队成员来自顶尖高校及科研院所,专业背景深厚,长期围绕纳米尺度下分子相互作用开展模拟计算工作。多年来从事纳米固-液界面与分子作用的模拟研究,在生物分子界面作用(小分子药物与蛋白、核酸结合),生物系统-纳米材料相互作用(生物分子与纳米材料吸附),纳米催化(电子转移过程)等领域有丰富的计算模拟工作经验。
研究所用的计算手段包括量化计算、分子动力学模拟、粗粒化模拟、分子对接等方法。对这些方法的相关软件也都十分精通,如量化软件Gaussian、VASP;分子动力学模拟软件NAMD、Gromacs、AMBER;粗粒化模型Martini、DPD;分子对接软件AutoDock、ZDOCK、PathDock等。熟练掌握Fortran,Shell,Python,TCL等多种编程语言,在数据分析过程中综合运用这些语言来编写处理脚本。
具体来说,研究过的目标体系主要有:水中纳米材料系统(自组装、力学响应、渗透和过滤体系)、生物分子与纳米材料系统(纳米颗粒表面分子吸附与交换、分子测序的机制分析、纳米材料或分子跨膜机制)、小分子与生物分子系统(小分子与生物大分子作用、信号通路药物虚拟筛选等)。
研究的内容包括配体–受体复合物体系的相互作用机制:相互作用模式、诱导契合效应、蛋白二级结构变化;预测活性小分子的结合自由能,阐明机理或指导结构改造;蛋白质结构预测,同源建模,优化蛋白结构。
膜蛋白分子动力学,各种磷脂膜的通透性研究,膜蛋白通道,纳米材料对磷脂膜的作用;拉伸分子动力学(SMD),蛋白解折叠研究,蛋白及碳纳米材料的弹性性质研究; 副本交换分子动力学(REMD),蛋白质结构转变研究等。
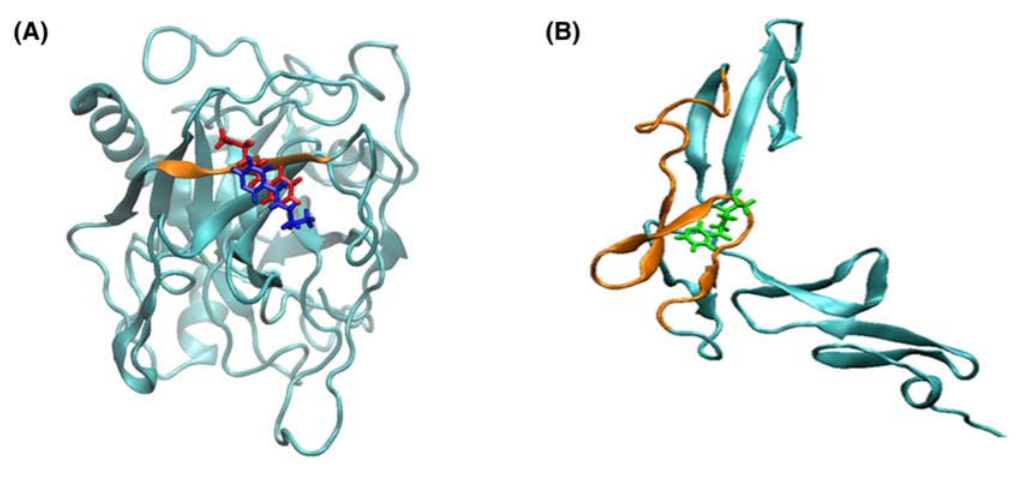
药物分子筛选

纳米器件模拟
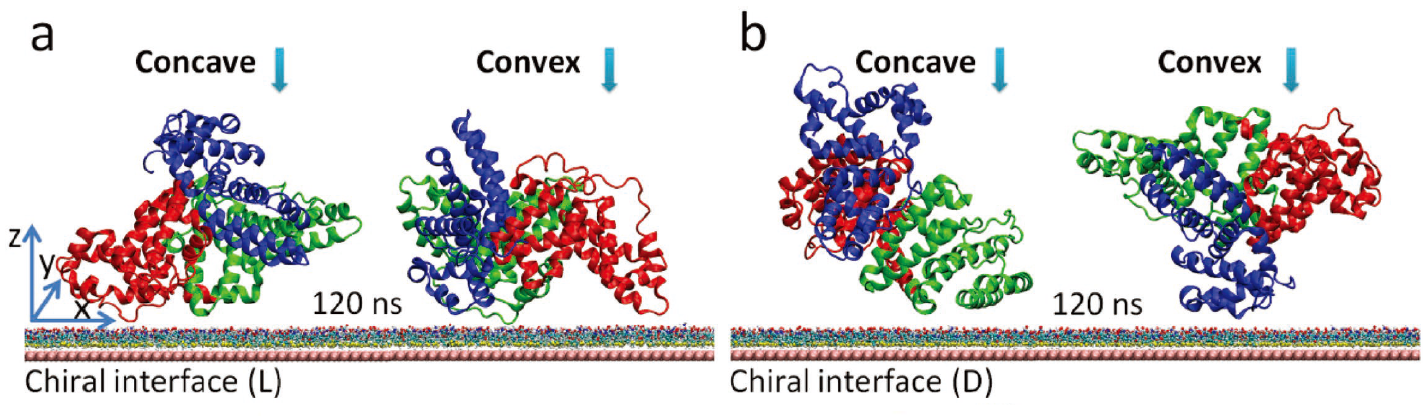
蛋白质与分子相互作用
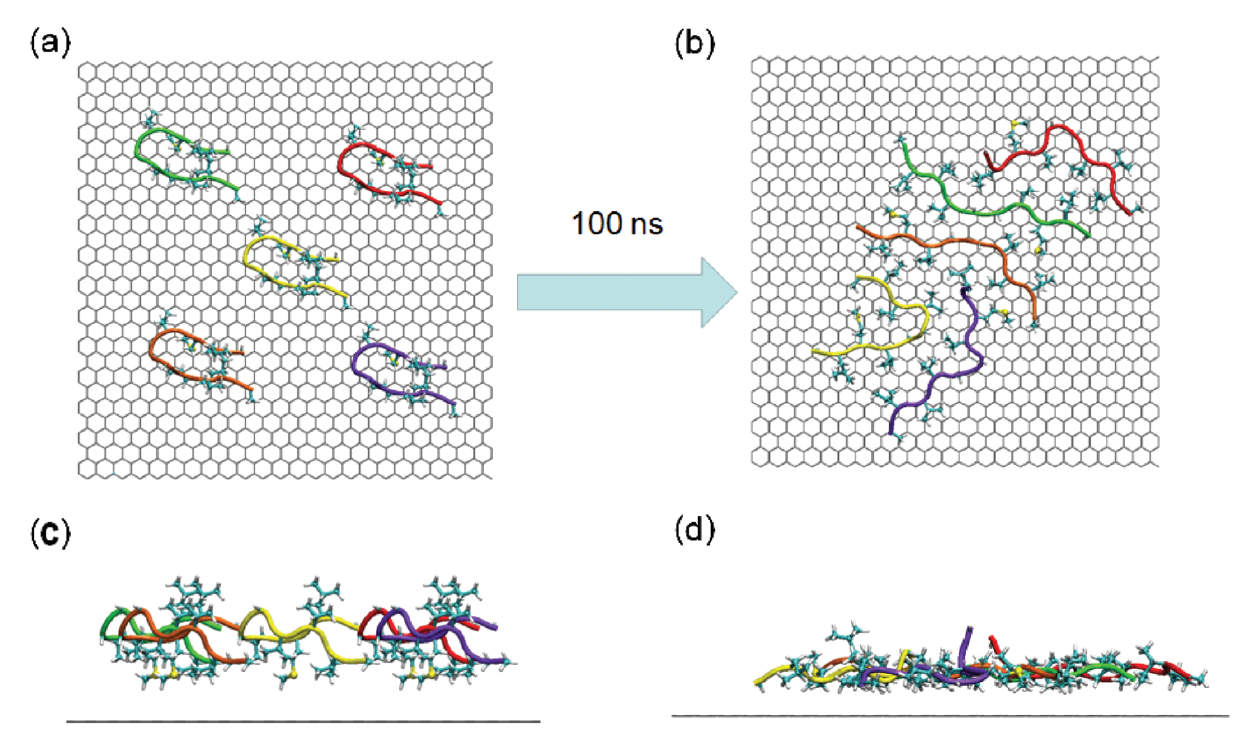
蛋白与纳米颗粒相互作用
用户相关成果在Nature Chemistry、PNAS、 JACS、 Angew. Chem.、 Nature Communications、Advanced Materials、Nano Letters、Environment Science & Technology 等期刊发表。





